





リリース
-
2026.06.25
共同プレスリリース
研究成果
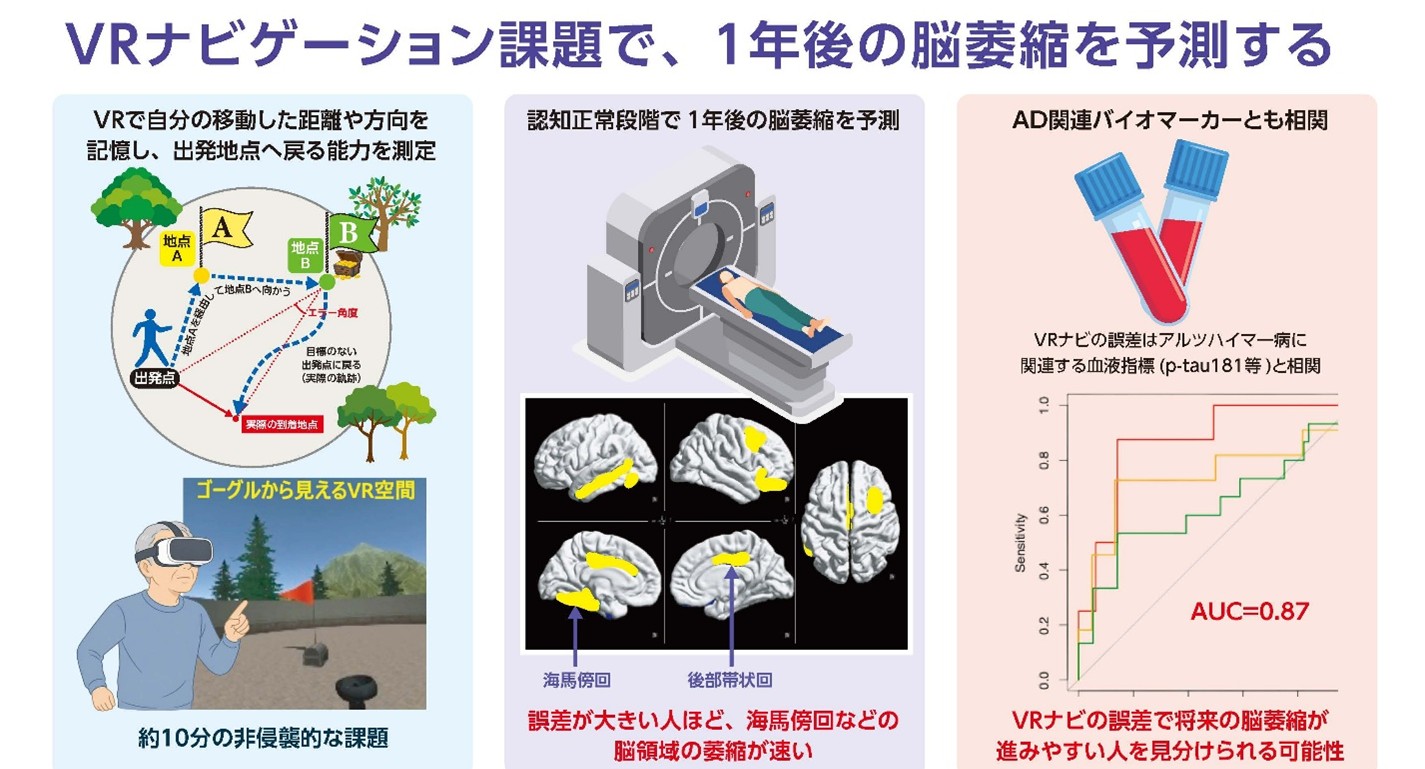
3D VRナビ課題の評価が1年後の脳萎縮を予測―認知機能が正常な段階で、認知症の超早期変化を捉える新規手法―
-

2026.06.24
プレスリリース
学習院大学とアクセルスペースの共同研究契約を締結―宇宙サステナビリティのルールメイキング戦略に関する共同研究―
-
2026.06.18
共同プレスリリース
研究成果
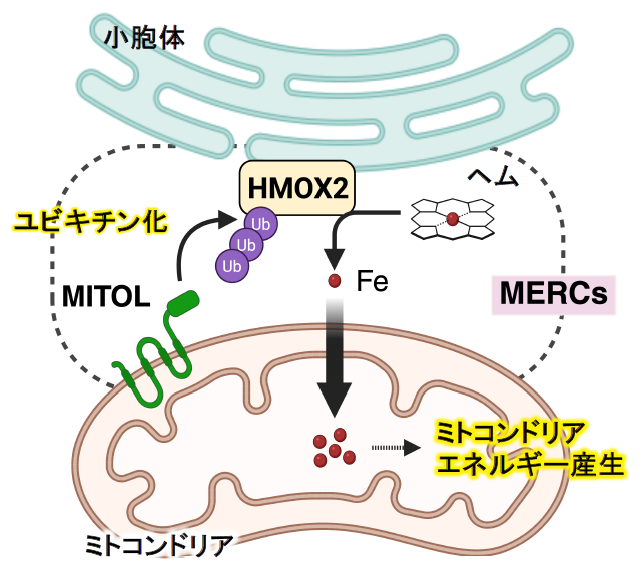
ミトコンドリアと小胞体の接触領域が「鉄供給ハブ」として働く―エネルギー産生を支える新たな鉄供給機構を発見―
-
2026.06.12
共同プレスリリース
研究成果
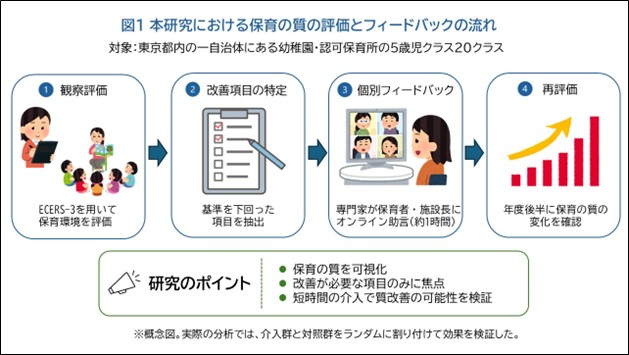
「保育の質」を可視化し、科学的視点に基づく改善へ―保育環境評価スケールに基づく専門性向上プログラムの効果を実証―
-
2026.06.11
共同プレスリリース
研究成果
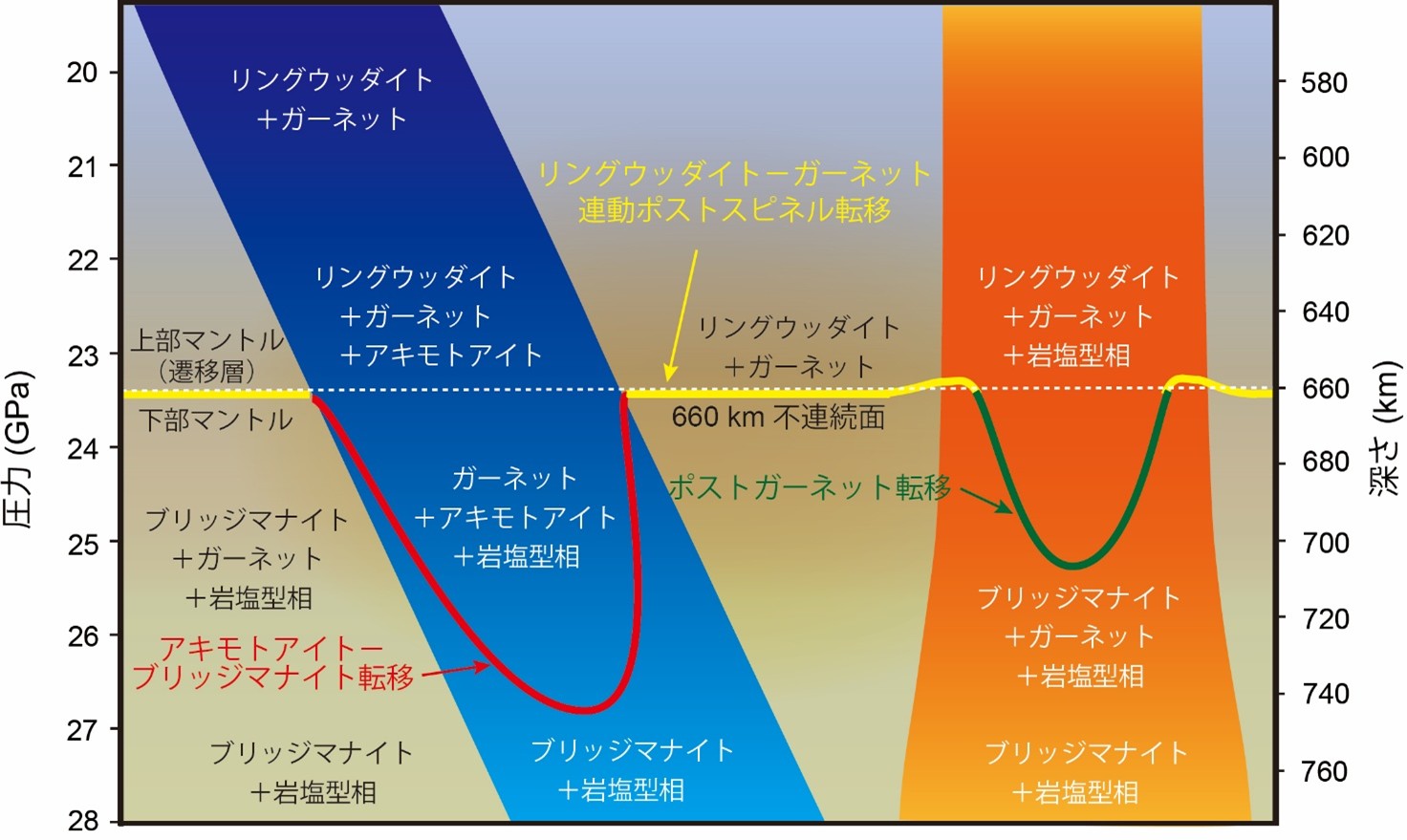
地球内部660 kmの境界形成はガーネットが支配していた
-

2026.05.22
リリース
研究成果
知識モデルにおけるunawareness表現の不可能性定理に関する新解釈を提示
-
2026.05.05
共同プレスリリース
研究成果
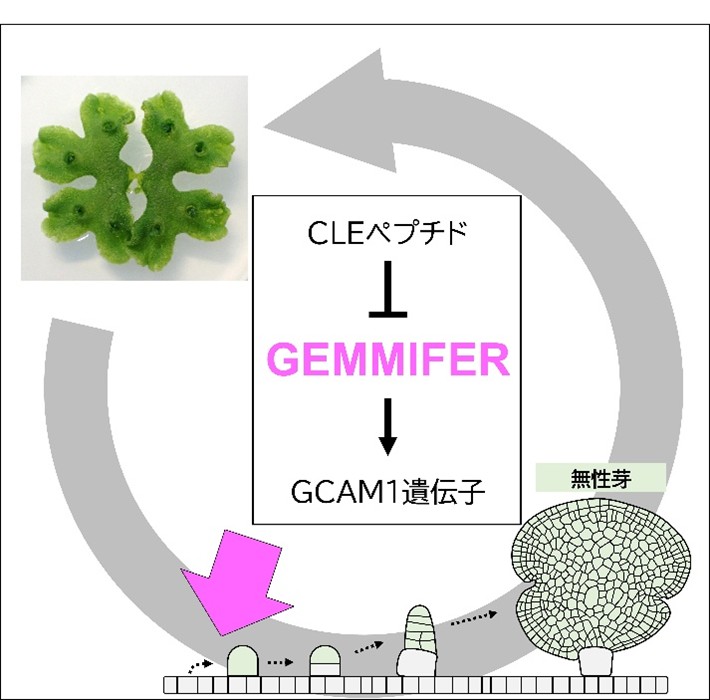
ゼニゴケのクローン繁殖を誘導する遺伝子を発見―農作物種を含むさまざまな植物への応用に期待―
-
2026.04.24
共同プレスリリース
研究成果
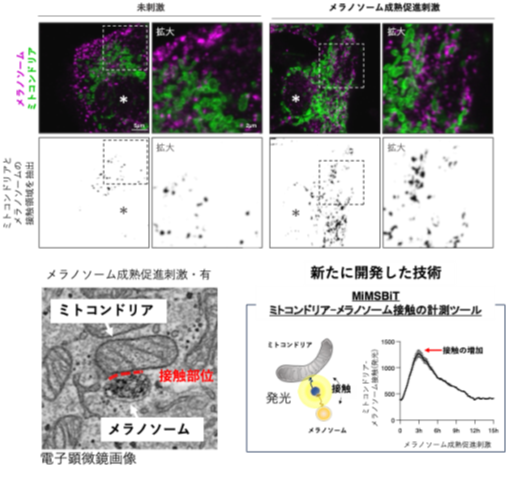
ミトコンドリアとメラノソームの接触がメラニン色素形成を制御―メラニン色素形成を支える細胞内機構を解明―
-

2026.04.22
共同プレスリリース
研究成果
老化で弱ったミトコンドリアを"増やして強くする"新規化合物を発見―加齢に伴う心機能低下を改善、栄養過多による寿命低下を抑制―
-
2026.04.07
研究成果
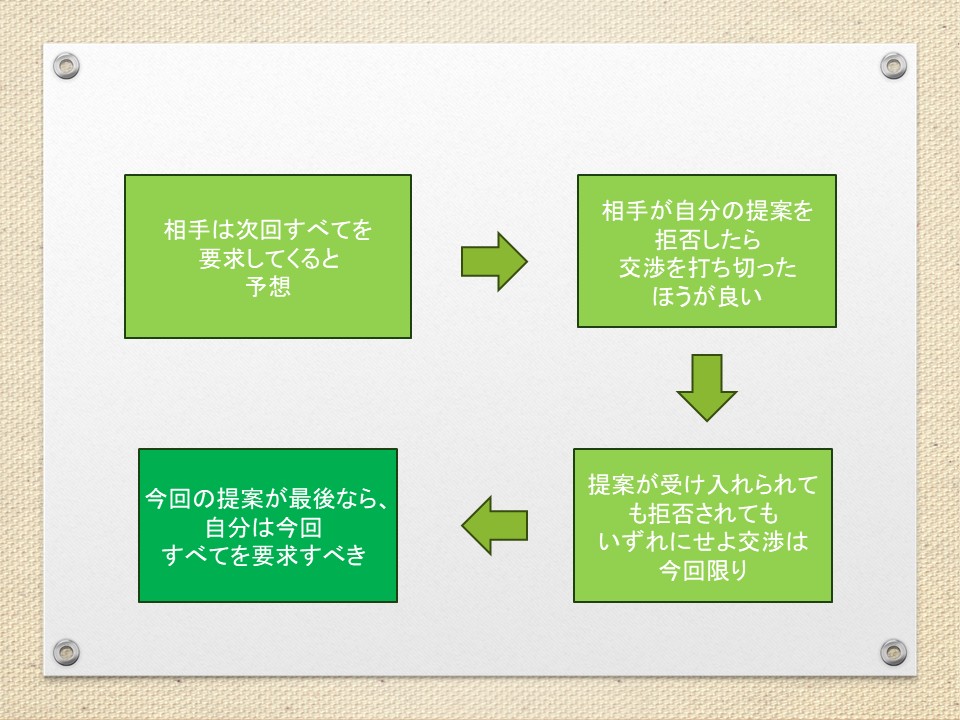
交渉において、提案を拒否したら打ち切るという脅し(最後通牒)が有効なのはどうしてか
-
2026.04.02
プレスリリース
研究成果
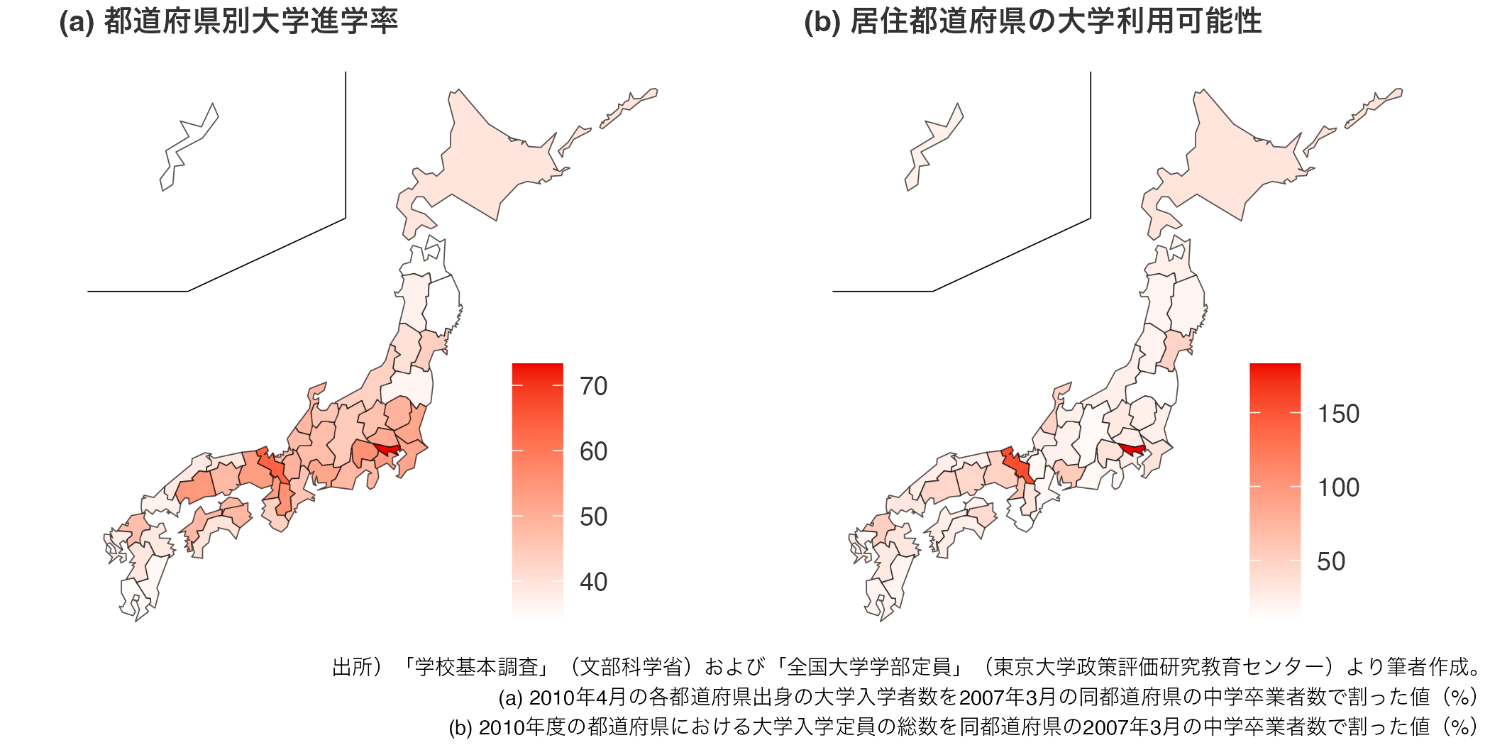
大学の偏在が進学率の地域間格差を拡大する 大学定員増加が地域の大学進学率に与える効果を実証
-
2026.03.24
プレスリリース
学習院大学発ベンチャー企業の株式会社マイトジェニックがミトコンドリア研究の新成分「マイトルビン」でダイドーグループと共同研究開始